
Molecular Dynamics Simulations (MD Simulations)
We have employed coarse-grained molecular approaches, including electrostatic potential (EP) calculations, docking, etc. as well as full-scale explicit molecular dynamics (MD) simulations to study the behavior of protein retention on ion-exchange and multimodal (MM) systems. The goal of these studies is to provide fundamental understanding of the nature of water-mediated protein-ligand interactions at a molecular level which can provide significant insight into the design of MM ligands, the roles of synergy and the modulation of selectivity using fluid phase modifiers (FPMs). This can further be used to explain, and indeed predict, the retention behavior of proteins on multimodal resins under a variety of solution conditions.
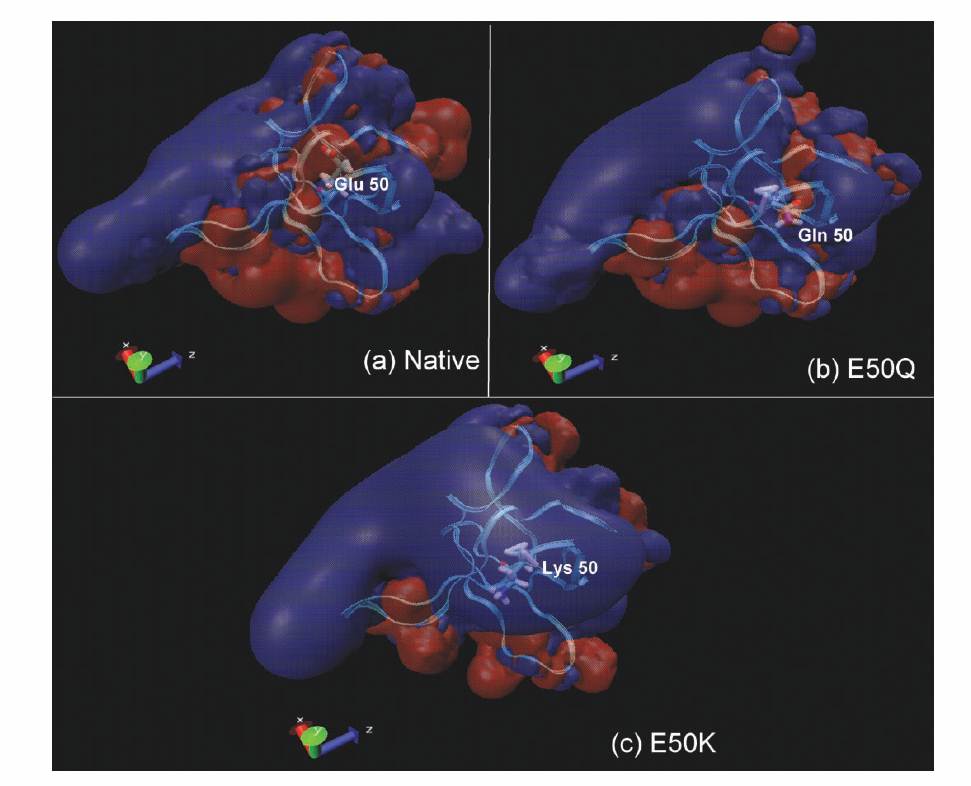
Electrostatic potential (EP) maps have been employed to understand the effect of protein surface topography, charge density and charge distribution on protein’s binding affinity and possible preferred binding orientations 1.
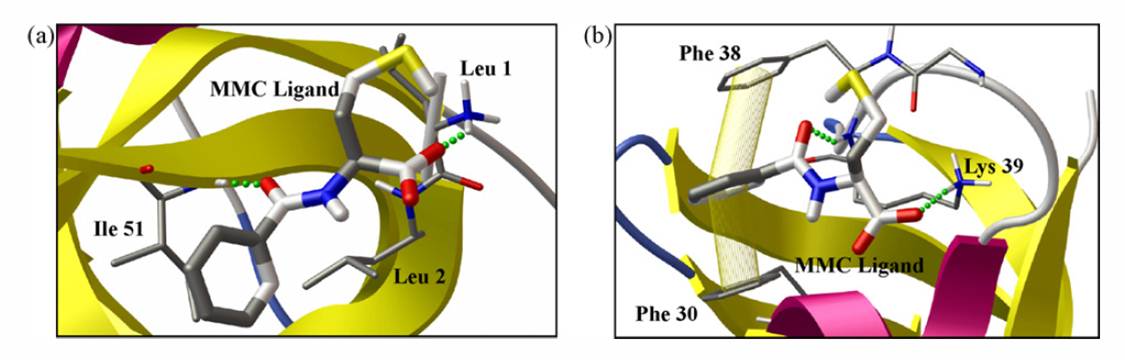
Molecular docking has been employed to examine the various interactions between the protein CspB and ligands in free solution 2, and to study the modes of interaction between MM ligand and ubiquitin 3.
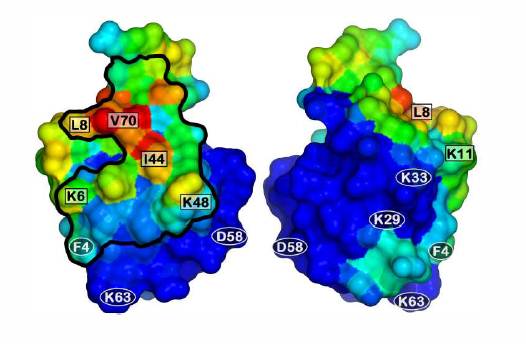
We have recently employed all-atom MD simulations of ubiquitin with multimodal ligands in free solution to identify binding preferences and to shed light on the “pseudo-affinity” nature of multimodal interactions. The results showed a preferred binding face on the protein, which corroborated the results obtained from NMR and chromatography experiments 4.
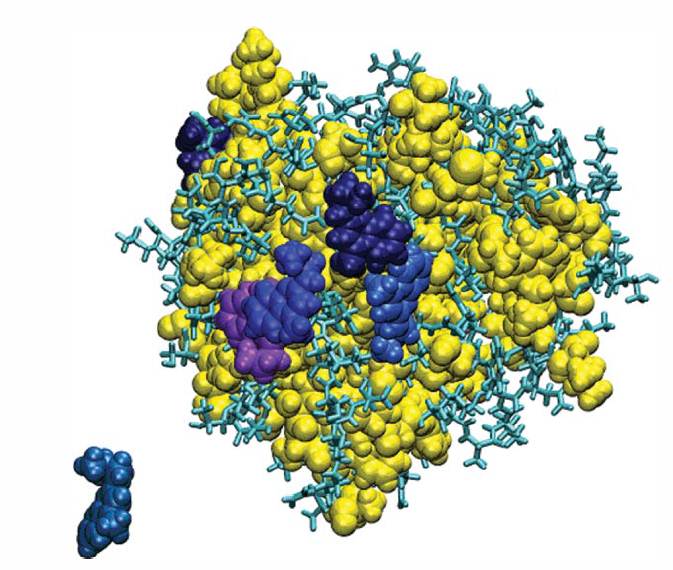
These results, in concert with data from benzene and acetate probes, also suggested that water-mediated electrostatic interactions may be more important for the localization of the MM ligand to the binding region with additional stability provided by a combination of local interactions such as hydrophobicity and hydrogen bonding. This hypothesis was also verified by performing all-atom MD simulations of K6E mutant of ubiquitin and it was observed that a reversal in charge at a binding site resulted in decreased binding even for hydrophobic residues 5.
MD simulations were also employed to confirm the selective binding between a protein and displacer as the mechanism by which chemically selective displacement occurs 6.
Selected References:
- Chung et al. Investigation of Protein Binding Affinity and Preferred Orientations in Ion Exchange Systems Using a Homologous Protein Library. Biotechnol. Bioeng. (2009) 102: 869-881.
- Chung et al. Investigation of protein binding affinity in multimodal chromatographic systems using a homologous protein library. J. Chromatogr. A 1217 (2010) 191-198
- Chung et al. Evaluation of protein adsorption and preferred binding regions in multimodal chromatography using NMR. PNAS (2010) 107 (39) 16811-16816.
- Freed el al. Molecular Simulations of Multimodal Ligand-Protein Binding: Elucidation of Binding Sites and Correlation with Experiments. J. Phys. Chem. B. (2011) 115 (45) 13320-13327.
- Holstein et al. Probing multimodal ligand binding regions on ubiquitin using nuclear magnetic resonance, chromatography, and molecular dynamics simulations. J. Chromatogr. A 1229 (2012) 113-20.
- Morrison et al. Mechanistic Studies of Displacer-Protein Binding in Chemically Selective Displacement Systems Using NMR and MD Simulations. Biotechno. Bioeng. (2009) 102: 1428-1437.